

Custom Search
|


|
Dermpath-India Pathology of Neurofibroma Dr Sampurna Roy MD 2022
|

 |
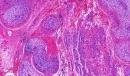
| Neurofibromas are the most
common benign peripheral nerve sheath tumour. These lesions
arise from endoneurium and the connective
tissues of peripheral nerve sheaths and
often appear as a soft, skin-coloured papule
or small subcutaneous nodule. Neurofibromas are comprised of Schwann
cells, fibroblasts, perineural cells, and mast cells in a variably myxoid
background. A mutation in the NF1 gene causes neurofibromas.
|
Solitary Neurofibroma: Solitary neurofibroma is a localized neurofibroma which is usually not associated with neurofibromatosis. Age: This lesion usually occurs between the ages of 20 and 30. Clinical presentation: Solitary neurofibromas are papular, nodular or pedunculated lesions and greyish white in colour. These painless, slow growing tumours are soft in consistency. Usually there is no evidence of secondary degenerative changes. Differential diagnosis: Schwannoma The tumour appears to originate within the endoneurium. Microscopic features: The lesion is nonencapsulated ( schwannoma is well encapsulated) and circumscribed. A grenz zone separates the lesion from the epidermis. The tumour is composed of interlacing bundles of elongated cells with wavy nuclei. Several small nerve fibres are also present. The tumour cells are set in a fibromyxoid backround. There are thin and thick collagen strands (shredded carrot collagen). Mast cells are present. Sometimes degenerative nuclear pleomorphism may be present (bizarre or atypical neurofibroma). Note: Presence of mitotic activity in neurofibroma is indicative of malignancy. (Mitosis is common in schwannoma). Variants: -Cutaneous Lipomatous Neurofibroma: -Myxoid neurofibroma - Abundant mucin is present in the matrix (S100 necessary to distinguish from intramuscular myxoma.) Differential diagnosis: - Myxoid DFSP -Collagenous neurofibroma - Thick collagen bundles are present in the matrix. -Epithelioid neurofibroma - The tumour cells are rounded with eosinophilic cytoplasm. -Granular neurofibroma - The cells contain granular PAS positive, diastase resistant cytoplasm. -Pigmented neurofibroma - Scattered tumour cells contain melanin pigment (S100 protein, HMB45 and Melan A positive). -Dendritic Cell Neurofibroma With Pseudorosettes: There is proliferation of all the elements of the peripheral nerve - Schwann cells, axons , perineural cells (plexiform type) and fibroblasts. Hence 30- 40% tumour cells are positive to S100 protein (in neurilemmoma 100% cells are positive). Axons are neurofilament positive. Factor XIIa positive and CD34 positive cells are also sometimes present.
Neurofibromatosis: Neurofibromatosis was described by von Recklinhausen in 1882. Diffuse Neurofibroma Age: Occurs between ages of 10-30 years. Site: Head and neck area. Clinical presentation: Plaque like elevation of the skin. On cut section, entire subcutis is thickened by firm greyish tissue. Microscopic features : Poorly defined lesion with diffuse replacement of the dermis and subcutis by neurofibromatous tissue. It diffusely infiltrate subcutaneous fat. It does not destroy but envelops the normal structure. The matrix of the lesion is uniform consisting of fine fibrillary collagen. The Schwann cells have shorter and more rounded contour in diffuse neurofibroma. Clusters of Meissner bodies may be present. Occasionally multinucleate giant cells may be present. Differential diagnosis: Dermatofibrosarcoma protruberance - More storiform pattern and CD34 positive and S100 protein negative. Plexiform Neurofibroma: Age: Usually occurs in children and young adults. Site: Head and neck region Clinical presentation: The expanded nerves form irregular, convoluted cords and nodules. Plexiform neurofibroma is associated with hyperpigmented skin, thickening of soft tissue and hypertrophy of bone. Microscopic features: Nodules of tortuous, expanded nerve branches cut in various planes of section. There is prominent myxoid change. In some cases cells spill out from the nerve into the soft tissue. In early stage, the affected nerve displays increase in endoneurial matrix. In the late stage the nerve fibres are replaced by proliferation of Schwann cells together with thick wavy collagen bundles. Presence of mitotic figures is indicative of malignancy. Immunohistochemistry:
Differential diagnosis: Plexiform Schwannoma
|
|
|
|
|
