

Custom Search
|


|
Dermpath-India Pathology of Pityriasis Lichenoides Dr Sampurna Roy MD 2022
|
|
Custom Search
|
|
Dermpath-India Pathology of Pityriasis Lichenoides Dr Sampurna Roy MD 2022
|
|
Pityriasis lichenoides
represents a group of inflammatory cutaneous diseases of unknown
etiology and with different clinical presentations.
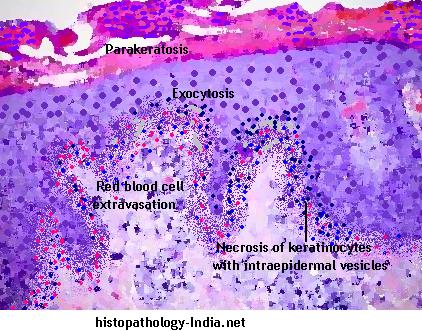
There are three subtypes of this disease : pityriasis lichenoides et varioliformis acuta (PLEVA), pityriasis lichenoides chronica (PLC) and febrile ulceronecrotic Mucha-Habermann disease. PLEVA and PLC are not distinct diseases but are different manifestations of the same process. PLC is six times more common than PLEVA. Pityriasis lichenoides affects children and young adults, peaking in the third decade of life. Men are three times more affected than women. -Clinical presentation: Pityriasis lichenoides et varioliformis acuta or PLEVA: Pityriasis lichenoides et varioliformis acuta or PLEVA consists of fairly extensive eruption, usually on the trunk and flexural areas of the extremities. There are acute papular lesions which rapidly form pseudovesicles and central necrosis. Individual lesions heal within few weeks with little or no scarring. Although individual lesions follow an acute course, the disorder is chronic, extending over several months or even years because of development of crops of new lesions. In a few patients some lesions increase in size to necrotic ulcers of 1 to 2 cm in diameter that heal with atrophic scarring. -Clinical presentation: Pityriasis lichenoides chronica or PLC : In pityriasis lichenoides chronica or PLC there are small, scaling, benign-appearing papules, mainly on the trunk. These lesions generally involute within 3 to 6 weeks. -Clinical presentation: Febrile Ulceronecrotic Mucha-Habermann disease. Febrile ulceronecrotic Mucha-Habermann disease is a severe variant of pityriasis lichenoides et varioliformis acuta characterized by the sudden onset of ulceronecrotic skin lesions and associated with high fever and systemic symptoms. Mucha separated the acute form of Pityriasis lichenoides from the chronic form of this disease in 1916. In 1966, Degos et al described an ulceronecrotic variant of acute Pityriasis lichenoides associated with fever, which was named Febrile Ulceronecrotic Mucha-Habermann disease. The diagnosis of this condition is suspected clinically and confirmed by histology. The microscopic features vary according to the phase, acute or chronic. The most common changes include: - Lymphocytic infiltrate with mixed neutrophils and histiocytes, - Exocytosis, - Parakeratosis, - Red blood cell extravasation and - Necrosis of keratinocytes with intraepidermal vesicles. Differential diagnosis: In immunohistochemistry the presence of CD30+ lymphoid cells and atypical lymphoid cells may occur. This may suggest the diagnosis of lymphomatoid papulosis, which is an important differential diagnosis of Pityriasis lichenoides. Differentiation between these two conditions is, important because patients with lymphomatoid papulosis, unlike those with pityriasis lichenoides, may develop systemic lymphoma. The presence of large CD30+ atypical lymphoid cells is the key feature of lymphomatoid papulosis. Other differences include the presence of large, atypical, non-lymphoid cells that may resemble Reed-Sternberg cells, many neutrophils, few lymphocytes, few or no necrotic keratinocytes, and little or no vacuolar degeneration of the basal layer, present in lymphomatoid papulosis unlike PLEVA . Clinically,the papules of lymphomatoid papulosis may develop into nodules, tumors, and large plaques (not seen in PLEVA). The cutaneous eruption tends to last much longer (sometimes even for years) than that of PLEVA. The differential diagnosis for PLEVA also includes arthropod bite reactions, varicella, Gianotti-Crosti syndrome, erythema multiforme, pityriasis rosea, guttate psoriasis, vasculitis, and secondary syphilis. Pathogenesis of Pityriasis lichenoides : a) Inflammatory reaction triggered by extrinsic antigens, such as: - Infectious agents (HIV, cytomegalovirus, Epstein-Barr virus, parvovirus B19, Toxoplasma gondii, Mycoplasma virus and Staphylococcus); - Drugs (hormone therapy with estrogen-progesterone, chemotherapeutics); and - Vaccine (MMR - mumps, measles and rubella). b) Lymphoproliferative origin. Some authors demonstrated the loss of mature antigen T cells CD2, CD3 and CD5 in PL, besides clonal proliferation of T cells in 50% of PLEVA cases (association with Hodgkin's lymphoma and lymphomatoid papulosis). c) Some authors demonstrated the presence of a component of immunocomplex-mediated vasculitis.
|
|
|
Visit:- Infectious Disease Online

Consultant Histopathologist (Kolkata - India)
|
![]()
Copyright © 2022 histopathology-india.net